Investigators:
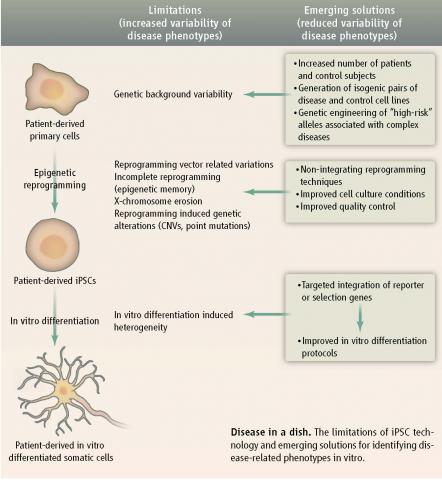
Dystroglycanopathies are a form of congenital muscular dystrophy (CMD) for which there is no known cure. It is an autosomal recessive disorder that is comparatively more common in Kuwait and the Middle East and is caused by the hypoglycosylation of a cell-surface glycoprotein, α-dystroglycan, leading to its reduced capacity to bind laminin in the extracellular matrix. Dystroglycanopathy patients present with progressive skeletal muscle weakness, degeneration, and often display a broad spectrum of neural phenotypes including mental retardation with and without structural brain abnormalities. A major challenge in CMD research and the development of CMD therapies is access to large quantities of hard-to-access, clinically relevant cell types (skeletal muscle and neural cells) for disease modeling and drug screening. Induced pluripotent stem cell (iPSC) technology offers the potential to develop such patient-specific model systems as iPSCs are derived from a patient’s somatic cells and have the potential to self-renew and differentiate into any cell type in the human body. In this pilot study, we will achieve the first steps needed to model this disease and screen for drugs in vitro:
1) collect fibroblasts and myoblasts from dystroglycanopathy patients in Kuwait with variety of disease-causing mutations, displaying a broad spectrum of phenotypes,
2) and generate induced pluripotent stem cells lines from these cells.
3) differentiating iPSCs into disease-relevant cell types and developing a cell-based, high content microscopy assay to screen for drugs based on the diseased cell’s reduced ability to bind laminin. Compounds identified in the screen to significantly increase the binding of laminin to iPSC-derived disease relevant cell types will be further studied to shed light on its mechanism of action and the complex relationship between the underlying genetic defect and the resulting diverse clinical phenotypes in dystroglycanopathy patients.
Project Outcome Report: Towards developing patient-specific induced pluripotent stem cell based assays for diseases modeling and drug screening